This command reads the parameters from the parameter
library, such as the CHARMM 22 parameter file for proteins with
all atoms [MacKerell et al., 1998]. This file contains the values for
bond lengths, angles,
dihedral angles, improper dihedral angles, and non-bonded interactions.
MODELLER relies on slightly modified CHARMM-22 parameters to reproduce the
protein geometry in the MODELLER environment. For example, for the
default non-hydrogen atoms model, the 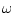 dihedral angle restraints
are stronger than the original CHARMM 22 values which apply to
the all-hydrogen model. For a sparse discussion of the parameter
library, see the FAQ Section 1.9, Question 17.
dihedral angle restraints
are stronger than the original CHARMM 22 values which apply to
the all-hydrogen model. For a sparse discussion of the parameter
library, see the FAQ Section 1.9, Question 17.
If ADD_PARAMETERS is on, the new parameters are added to the existing
parameter list, otherwise the contents of the new parameter file
replaces the old one.
The filename for the library is DIRECTORY/FILE.