Analysis of Sequence and Structure Relationships
in the Calycin Protein Superfamily
Darren R Flower
Department of Physical Chemistry and BioAnalysis
Astra Charnwood,
Bakewell Road, Loughborough, Leicestershire, UK, LE11 ORH
Introduction
The Calycin protein superfamily [1,2], an example of a so-called "structural
superfamily" where obvious similarities of three-dimensional structure are not easily
discernible at the sequence level, is composed of three distinct families of ligand binding
proteins. The Lipocalins form a diverse and rapidly burgeoning family of predominantly
small secreted proteins; members of the family exhibit high affinity and selectivity for
hydrophobic molecules and have been generally characterized as extracellular transport
proteins; in this respect, the family is typified by long-established members such as retinol
binding protein (RBP), which transports retinol through the blood to target cells. The Fatty
Acid-Binding Proteins, or FABPs, another recently identified family of small, almost
exclusively intracellular proteins, which again bind hydrophobic molecules, are thought to be
involved in lipid metabolism. These proteins are typified by cellular retinol binding protein
(cRBP), which is believed to function in the intracellular trafficking of retinol. The Avidins,
such as streptavidin, a small soluble protein from the bacteria Streptomyces avidinii, and its
homologue hen egg-white avidin, are proteins with a remarkable affinity for the vitamin biotin,
as well as other hydrophobic ligands, and have found important applications in biotechnology.
Structural Relationships
Crystallographic studies of several lipocalins have shown them to possess a conserved
folding pattern dominated by an 8 stranded calyx- or cup-shaped antiparallel b-barrel which
encloses an internal ligand binding site [1]. The 8 strands of the barrel (labelled A to H) are linked
by a succession of +1 connections, which may be formally regarded as b-hairpin loops
(labelled L1 to L7), giving the simplest possible topology for a closed b-sheet. Loops L2 to L7
are typical of short b-hairpins, while loop L1 is a large W loop which forms a lid folded back to
cap the internal ligand binding site. This loop varies in conformation between different
lipocalins but maintains its overall shape, size, and position. A short 310 helix, directly before
strand A, helps to close off the other end of the barrel. Beyond the last strand of the b-barrel, is
an a-helix which folds back to pack parallel with strands G and H.
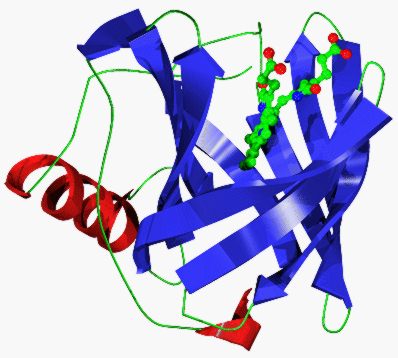
The crystal structures of several FABPs comprise a well conserved 10 stranded
antiparallel b-barrel, again with a repeated +1 topology, although it is not continuously
hydrogen bonded, with a wide discontinuity between strands D & E1. A short 310 helix,
leading into strand A, partly closes off one end of the barrel. In the FABPs the region between
strands A and B form two short but well defined a-helices which pack onto the top of the lipid
binding site. If considered as a loop connecting two strands then this feature is very similar to
loop L1 of the lipocalin fold (also a large loop folded back to cap the internal cavity).
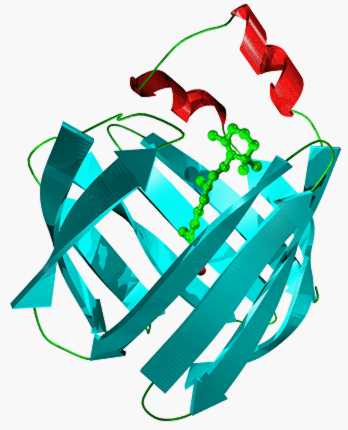
The avidin fold is dominated by 8 b-strands which form an antiparallel b-barrel
enclosing the internal biotin binding site [2]. These 8 strands are linked by a succession of seven
+1 connections (L1 to L7). These loops, although they vary in size and conformation, are all
typical of short b-hairpins. In the lipocalins and FABPs, loop L1 is a large, atypical W-loop,
while in the avidins loop L1 is, by contrast, a short, typical b-hairpin. A short 310 helix,
directly before strand A, helps to close off one end of the barrel.
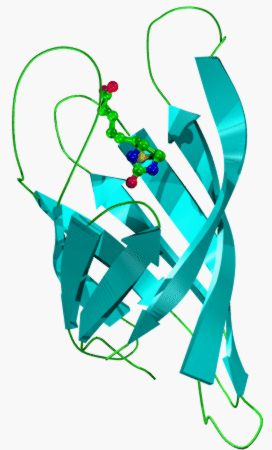
The structures of a lipocalin, human retinol binding protein, and a FABP, rat intestinal
fatty acid-binding protein (I-FABP), can be superimposed onto that of streptavidin [2]. The
topology of all three structures is very similar: each form antiparallel b-barrels with repeated +1
connections, although the FABP barrel is more flattened or elliptical than that of the lipocalins
and the avidin barrel is both more circular and compact, in cross-section, than that of the
lipocalins. The three structures can be equivalenced so that the initial four and final two strands
of each barrel match to form contiguous structurally conserved regions (SCRs), with little or
no correspondence between strands from the centre of their closed b-sheets or between any
intervening loop regions. Such structural alignments show that the first SCR of the calycin
common core has the same unusual conformation (a 310 helix leading into a b-strand) and
location within the fold of the lipocalins, FABPs, and avidins. It contains within it a highly
conserved sequence motif common to all three families.
Sequence relationships
In recent years, the apparent size and diversity of both the lipocalin and FABP families
has grown significantly, as new members are proposed on the basis of sequence similarity, to
encompass a large number of proteins. In contrast, the number of extant avidin sequences
remains small.
The common core of the lipocalin fold is dominated by three large SCRs, which bear
characteristic, conserved sequence motifs. Using possession of these three key motifs as a
definition of family membership allows identification of the largest self-consistent subset
within the whole set of related sequences [1,3]. This analysis shows the lipocalin protein family
to be composed of a main core group of quite closely related proteins: the kernal lipocalins,
and a smaller number of more structurally, and possibly functionally, divergent sequences
(such as the a-1-acid glycoproteins, odour binding proteins, and Von Ebner's gland proteins):
the outlier lipocalins. It may be misleading to say a protein is a lipocalin, with all that that
implies for shared structure and function (such as cell surface receptor binding), because it has
some global sequence similarity to an established member of the family; rather, for certain
inferences, particularly structural ones, to be drawn with confidence from such sequence
similarity, all three key characteristic motifs must also be present.
Moreover, within the lipocalin fold, residues from both SCR1 and SCR2 make several
specific contacts with a conserved feature near the end of SCR3: the terminal residue of strand
G. This is an arginine in most lipocalins and a lysine in some others. The end of this side chain
makes hydrogen bonds to a ring of acceptors, main chain carbonyls derived from either the
310-helix leading into strand A or from the apex of loop L6, and its side chain packs, in an
extended conformation, against the totally conserved tryptophan at the start of strand A. These
highly conserved residues form a characteristic patch on the surface of the proteins [1].
A similar arrangement of side chains interactions - a tryptophan, from the first strand of
the b-barrel, packing against an arginine from near the end of the last - is also observed in the
FABP structures, although this interaction is not as extensive as that seen in the lipocalins, for
example there is no equivalent to the SCR2 interaction.
Examination of the available structures of the chicken avidin and streptavidin reveals
that a very similar arrangement of interacting residues is also, seemingly, a conserved feature
of the third member family of the calycins.
It is clear then that in all three families comprising the calycin superfamily, a similar
pattern of packing between characteristically conserved residues is apparent. An arginine or
lysine, able to form a number of potential hydrogen bonds with the main chain carbonyls of a
short 310 helix, packs across a conserved tryptophan [4]. This conservation of particular residues
and their interactions across the three member families suggests that this structural signature,
seemingly characteristic of the calycin superfamily, might also correspond to a characteristic
sequence signature. To test this contention, two short sequence motifs, including the residues
participating in these key interactions, were identified within the first and last SCRs of the
overall calycin common core and used as the basis for a regular expression summarising the
different residue types observed at each alignment position. Combining these with motif
separations gives a search pattern whose performance compares favourably with those of much
more sophisticated search methods [5,6].
The implications of this work are clear, a structural signature characteristic of almost the
whole calycin family corresponds to a sequence pattern able to identify a significant subset of
their sequences. These conserved side chain-side chain and side chain-main chain interactions
act to pin together the two ends of the calycin b-barrel. But what is the role of this key
structural motif ? It may stabilize the overall protein structure and help maintain the b-barrel
fold, it may guide its formation as a key interaction in the folding pathway or it may have some
functional role, perhaps as a protein-protein recognition site. Alternatively, in these present day
structures, the sequences that give rise to the individual folding patterns of each family may
have evolved stability independent of this residue packing and all we see, in this interaction, is
an echo of an important past interaction; still favourable and / or important but no longer
essential: an evolutionary relic or leftover of the now distant common calycin ancestor protein.
Discussion
Beyond an obvious similarity of function, the triumvirate of protein families composing
the calycin superfamily is characterized by a similar overall folding pattern (an antiparallel b-
barrel, with a repeated +1 topology, possessed of an internal ligand binding site), within which
large amounts of the lipocalin, FABP, and streptavidin structures can be meaningfully
equivalenced to form a number of significant discrete structurally conserved regions [1,2]. This is
indicative of a close relationship between the characteristic folds of these families. The first
SCR, within the common core characteristic of the three families, corresponds to an unusual
structural feature (a short 310 helix leading into a b-strand, the first of the barrel), which is
conserved in its conformation and location within the fold of the lipocalins, FABPs, and
avidins and displays a characteristic pattern of sequence conservation. Notwithstanding the lack
of significant global sequence similarity between any of these three groups, this motif is
present in virtually all of the calycins.
Protein structures are rather better conserved than their sequences and in extreme cases
close relationships are not apparent from the analysis of sequence alone: two views of this
phenomenon have emerged. One maintains that although structural propinquity has been
retained, evolutionary divergence from a common ancestor has occurred beyond which any
significant sequence similarity remains. The other proposes a convergent mechanism whereby
structures resemble each by chance because of the limited number of thermodynamically stable
folds. In the calycins, the retention of characteristic and conserved sequence signatures
corresponding to a common pattern of side chain packing argues for the former explanation and
supports the conjecture that the lipocalins, FABPs, and avidins share an evolutionary
relationship [4].
References
1 Flower, D.R., North, A.C.T., and Attwood, T.K. (1993), Structural and sequence relationships in the
lipocalins and related proteins, Prot. Sci. 2, 753-761.
2 Flower, D.R., (1993), Structural relationship of streptavidin to the calycin protein superfamily, FEBS
Letters, 333, 99-102.
3 Flower, D.R., North, A.C.T., and Attwood, T.K. (1991), Mouse oncogene protein 24p3 is a member of the
lipocalin protein family, Bioch.Biophys.Res.Comm. 180, 65-74.
4 Flower, D. R. (1995), A Structural Signature characteristic of the Lipocalin Protein Family, Prot. Pep. Lett.
2, 341-346.
5 Sansom, C.E., North, A.C.T., and Sawyer, L. (1994), Bioch. Biophys. Acta 1208, 247-255.
6 Johnson, M.S., Overington, J.P, and Blundell, T.L. (1993), J. Mol. Biol. 231, 735-752.
Database Searching for Inhibitors of Secretory Phospholipase A2 using CLIX
N P Tomkinson, D R Flower, D P Marriott, N P Gensmantel,
and S Connolly *
Department of Physical Chemistry and BioAnalysis and * Medicinal Chemistry
Astra Charnwood,
Bakewell Road, Loughborough, Leicestershire, UK, LE11 ORH
Phospholipase A2 (PLA2) catalyses the hydrolysis of membrane phospholipids to yield a
cascade of lipid-derived mediators of inflammation including arachadonic acid and it's metabolites
the eicosanoid prostaglandins and leukotrienes.
As part of an ongoing program to devise specific PLA2 inhibitors as anti-inflammatory
agents we have used CLIX [1] to find potential new ligands for secretory PLA2 (sPLA2) from the
Cambridge Structural Database (CSD) [2]. CLIX represents a novel approach to database searching.
Rather than using a simple pharmacophore to define a search, CLIX uses the protein structure to
define an array of target sites that are used to find complementary structures in the database. The
hits were scored using GRID version 10.0 [3].
In order to be confident that the software was working in our hands we used it to
reproduce the crystal structure of human sPLA2 complexed with a transition state analogue, L-1-
O-octyl-2-heptylphosphonyl-sn-glycero-3-phosphoethanolamine [4]. The ligand was reduced in size
and complexity in order to provide a more rigorous test of the method. To include the full structure
would almost certainly preclude alternative orientations of the ligand within the active site and
therefore provide an unrealistic estimate of the predictive power of the software. A CSD format
database containing this one structure was formed and searched using CLIX.
Having satisfied ourselves that the program is producing sensible results we then searched
the CSD. Steric complementarity of the
first hit is good, and the oxygen of one of the carboxylate groups is placed adjacent to the calcium
ion. The interaction energy used for scoring will be dominated by the presence of the calcium ion.
This is why all the hits are carboxylic acids.
CLIX has successfully reproduced the inhibitor-ligand crystal structure and retrieved
convincing hits that provide new starting points for synthesis. We intend to convert other
commercial and in-house databases into CLIX format and also to convert target points into a
pharmacophore for conventional database searching. An obvious refinement of this whole
approach would be to construct a conformational database first.
References
1 Lawrence,M.C. and Davis, P.C., PROTEINS:Structure,Function & Genetics (1992), 12, 31-41.
2 Cambridge Structural Database System, Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge,
CB2 1EZ.
3 Goodford, P.J., J.Med.Chem. (1985), 28, 849-857.
4 Scott, D.L., White, S.P., Browning, J.L., Rosa, J.J., Gelb, M.H. and Sigler, P.B., Science (1991), 254, 1007-
1010.