
Thanks Ben,
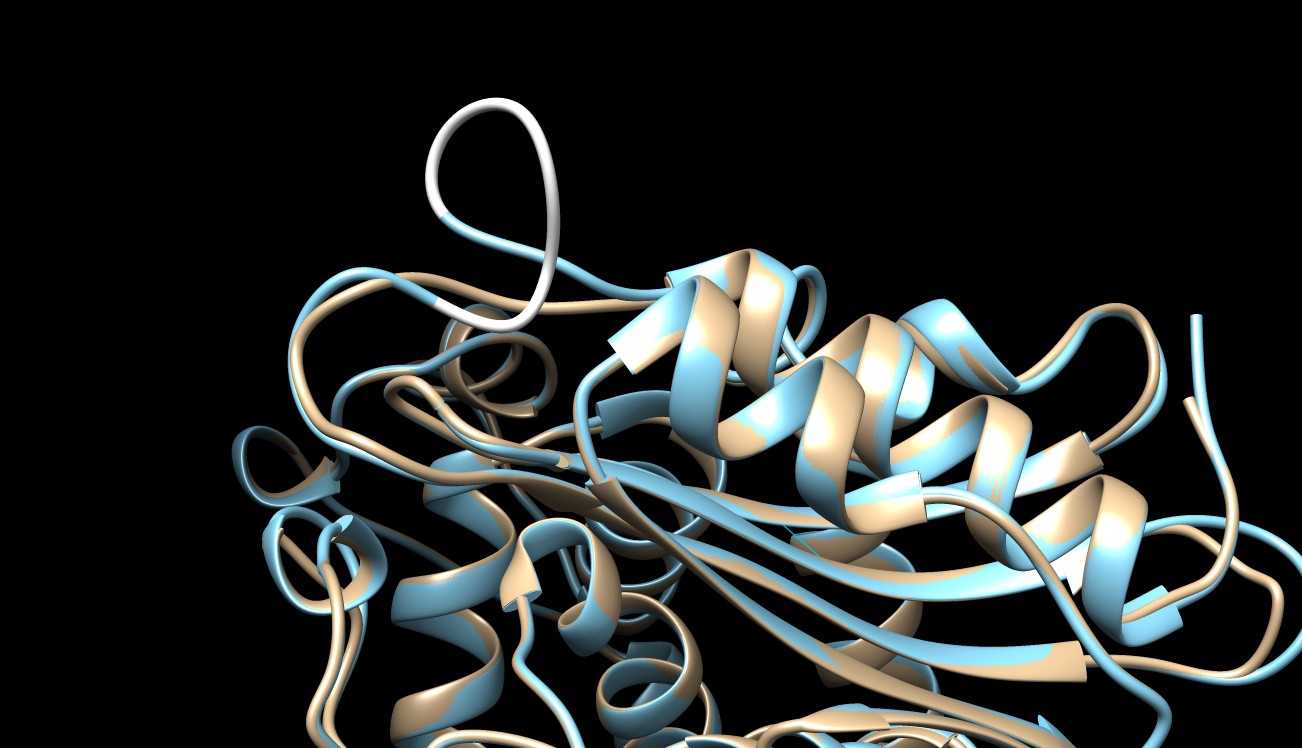
is there a way to avoid that an insertion from becoming a curious loop like the one shown at the top in the figure?
I’m trying to relax the system using MD but maybe I can avoid it during modeling.
Best,
Marco
Il 22/10/2024 01:26, Modeller Caretaker ha scritto: > On 10/21/24 2:28 AM, Marco Sette via modeller_usage wrote: >> I have obtained five models of a protein with Modeller and I am >> analyzing them in relation to the wt. There are of course small >> deviations, and such differences affect some physical properties. In >> your opinion, should I analyze the separate models or I can take the >> average structure? In NMR, people often use an average structure so I >> wonder if it makes sense in the modelling field. > > Normally we look at the individual models. If you do want to average > the structures, I would recommend you at least relax the final > structure. One way to do that is to use AutoModel.cluster() as per > https://urldefense.com/v3/__https://salilab.org/modeller/10.5/manual/node71.... > which does a basic conjugate gradients minimization. > > Ben Webb, Modeller Caretaker
{kind=link}