- ... alignment.2.1
- If the residue type is defined in
'modlib/restyp.lib' you can use the 1-letter code that is specified there,
but if in doubt use '.', since that matches everything.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... assigned2.2
- After
uppercase letters A-Z are used, chain IDs 0 through 9 are assigned, then
lowercase letters a-z. If your protein contains more than 62 chains, the
remaining chains are given no IDs.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... restraints2.3
- The
restraints include bond, angle and dihedral angle restraints.
The SG — SG atom pair also becomes an excluded atom pair that is not checked
for an atom-atom overlap. The
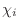 dihedral angle restraints will depend
on the conformation of the equivalent disulfides in the template structure,
as described in [Šali & Overington, 1994].
dihedral angle restraints will depend
on the conformation of the equivalent disulfides in the template structure,
as described in [Šali & Overington, 1994].
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
exception5.1
- MODELLER can raise the following Python exceptions:
ZeroDivisionError, IOError, MemoryError, EOFError,
TypeError, NotImplementedError, UnicodeError, IndexError, and ValueError.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... name5.2
- All MODELLER Python classes are
named using CamelCase, such as Model, Alignment, or
AutoModel. Previous versions of MODELLER used lowercase names,
such as model, alignment, or automodel. These lowercase names
are still available but are deprecated.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... special.5.3
- On Unix/Linux systems, MODELLER assumes that your filesystem
also stores filenames in UTF-8. This is usually the case on modern systems;
however, you can change the encoding by setting the G_FILENAME_ENCODING
environment variable.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... numbers6.1
- for mmCIF format,
author-provided residue numbers (_atom_site.auth_seq_id) are used if
available rather than the canonical sequence id
(_atom_site.label_seq_id)
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... library6.2
- If no atoms in the first
chain have coordinates, internal coordinate generation is seeded by placing the
first atom at the origin, the second along the x axis, and the third in the xy
plane. If no atoms in subsequent chains have coordinates, the first atom is
placed 2 angstroms along each of the x, y and z axes from the last atom in the
previous chain, and the second and third atoms placed in the same way as for
the first chain.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... communication)6.3
-
Fractional surface area of a residue X is given as the calculated surface area
divided by that of the residue in an extended tripeptide Ala-X-Ala
conformation.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
scores6.4
- The mean and standard deviation of the DOPE score of a given
protein is estimated from its sequence. The mean score of a random protein
conformation is estimated by a weighted sum of protein composition over the
20 standard amino acid residue types, where each weight corresponds to the
expected change in the score by inserting a specific type of amino acid
residue. The weights are estimated from a separate training set of
1,686,320 models generated by MODPIPE.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
6.5
- The target distances were all obtained from a regular α-helix in
one of the high-resolution myoglobin structures.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...http://www.ks.uiuc.edu/Research/vmd/.
6.6
- Note that binary trajectory files are machine dependent; it is up
to the visualization software to do any necessary byte-swapping.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... files6.7
- This won't work in combination
with write_whole_pdb = False for structures
that were added to the alignment with Alignment.append_model(), since
such inputs may not have an atom file to extract the directory from. In this
case the outputs will end up in the current directory.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
out6.8
- Any structures that were added with Alignment.append_model()
will need to have their corresponding atom files available, so that the
originals can be reread at this point.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... files6.9
- This won't work in combination
with write_whole_pdb = False for structures
that were added to the alignment with Alignment.append_model(), since
such inputs may not have an atom file to extract the directory from. In this
case the outputs will end up in the current directory.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
out6.10
- Any structures that were added with Alignment.append_model()
will need to have their corresponding atom files available, so that the
originals can be reread at this point.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... model6.11
- residue numbers in mmCIF refer to
author-provided values, if available (e.g. atom_site.auth_seq_id),
otherwise to label_seq_id.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...'m'6.12
- Note that 'm.chains['A'].residues['10']' in most cases is
the same as 'm.residues['10:A']'. However, if there are multiple chains in
the model called 'A' the first syntax will return residue 10 in the first chain
called A (and so will fail if that chain does not contain this residue) whereas
the second will look for the first residue numbered 10 in any of the A chains.
(Generally speaking, you should avoid having duplicate chain IDs or residue
numbers!)
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... codeB.1
-
for PDB format, this is a 1-letter chain ID; for mmCIF format, this is the
author-provided chain/asym ID (_atom_site.auth_asym_id) if available,
otherwise the canonical asym ID (_atom_site.label_asym_id), both of
which can be multiple characters in length
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.