- ... alignment.2.1
- If the residue type is defined in
'modlib/restyp.lib' you can use the 1-letter code that is specified there,
but if in doubt use '.', since that matches everything.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... assigned2.2
- After
uppercase letters A-Z are used, chain IDs 0 through 9 are assigned, then
lowercase letters a-z. If your protein contains more than 62 chains, the
remaining chains are given no IDs.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... restraints2.3
- The
restraints include bond, angle and dihedral angle restraints.
The SG -- SG atom pair also becomes an excluded atom pair that is not checked
for an atom-atom overlap. The
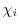 dihedral angle restraints will depend
on the conformation of the equivalent disulfides in the template structure,
as described in [Šali & Overington, 1994].
dihedral angle restraints will depend
on the conformation of the equivalent disulfides in the template structure,
as described in [Šali & Overington, 1994].
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
exception5.1
- MODELLER can raise the following Python exceptions:
ZeroDivisionError, IOError, MemoryError, EOFError,
TypeError, NotImplementedError, UnicodeError, IndexError, and ValueError.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... special.5.2
- On Unix/Linux systems, MODELLER assumes that your filesystem
also stores filenames in UTF-8. This is usually the case on modern systems;
however, you can change the encoding by setting the G_FILENAME_ENCODING
environment variable.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ... communication)6.1
-
Fractional surface area of a residue X is given as the calculated surface area
divided by that of the residue in an extended tripeptide Ala-X-Ala
conformation.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
scores6.2
- The mean and standard deviation of the DOPE score of a given
protein is estimated from its sequence. The mean score of a random protein
conformation is estimated by a weighted sum of protein composition over the
20 standard amino acid residue types, where each weight corresponds to the
expected change in the score by inserting a specific type of amino acid
residue. The weights are estimated from a separate training set of
1,686,320 models generated by MODPIPE.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...
6.3
- The target distances were all obtained from a regular
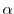 -helix in
one of the high-resolution myoglobin structures.
-helix in
one of the high-resolution myoglobin structures.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
- ...http://www.ks.uiuc.edu/Research/vmd/.
6.4
- Note that binary trajectory files are machine dependent; it is up
to the visualization software to do any necessary byte-swapping.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.