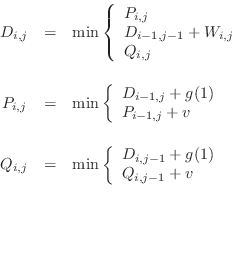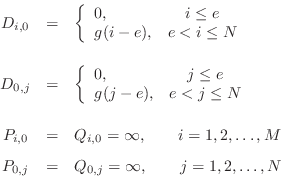Next: Variable gap penalty Up: Dynamic programming for sequence Previous: Dynamic programming for sequence Contents Index
The residue by residue scores ![]() can be used directly in the
sequence alignment algorithm of Needleman & Wunsch [Needleman & Wunsch, 1970]
to obtain the comparison of two protein sequences or structures. The only
difference between the two types of comparison is in the type
of the comparison matrix. In the case of sequence, the amino acid
substitution matrix is used. In the case of 3D structure, the Euclidean
distance (or some function of it) between two equivalent atoms in
the current optimal superposition is used [Šali & Blundell, 1990].
can be used directly in the
sequence alignment algorithm of Needleman & Wunsch [Needleman & Wunsch, 1970]
to obtain the comparison of two protein sequences or structures. The only
difference between the two types of comparison is in the type
of the comparison matrix. In the case of sequence, the amino acid
substitution matrix is used. In the case of 3D structure, the Euclidean
distance (or some function of it) between two equivalent atoms in
the current optimal superposition is used [Šali & Blundell, 1990].
The problem of the optimal alignment of two sequences as addressed by
the algorithm of Needleman & Wunsch is as follows. We are given two
sequences of elements and an ![]() times
times ![]() score matrix
score matrix ![]() where
where
![]() and
and ![]() are the numbers of elements in the first and second
sequence. The scoring matrix is composed of scores
are the numbers of elements in the first and second
sequence. The scoring matrix is composed of scores ![]() describing
differences between elements
describing
differences between elements ![]() and
and ![]() from the first and second
sequence respectively. The goal is to obtain an optimal set of
equivalences that match elements of the first sequence to the elements
of the second sequence. The equivalence assignments are subject to the
following “progression rule”: for elements
from the first and second
sequence respectively. The goal is to obtain an optimal set of
equivalences that match elements of the first sequence to the elements
of the second sequence. The equivalence assignments are subject to the
following “progression rule”: for elements ![]() and
and ![]() from the
first sequence and elements
from the
first sequence and elements ![]() and
and ![]() from the second sequence, if
element
from the second sequence, if
element ![]() is equivalenced to element
is equivalenced to element ![]() , if element
, if element ![]() is
equivalenced to element
is
equivalenced to element ![]() and if
and if ![]() is greater than
is greater than ![]() ,
, ![]() must also be greater than
must also be greater than ![]() . The optimal set of equivalences is the
one with the smallest alignment score. The alignment score is a sum of
scores corresponding to matched elements, also increased for
occurrences of non-equivalenced elements (ie gaps). For a detailed
discussion of this and related problems see [Sankoff & Kruskal, 1983].
. The optimal set of equivalences is the
one with the smallest alignment score. The alignment score is a sum of
scores corresponding to matched elements, also increased for
occurrences of non-equivalenced elements (ie gaps). For a detailed
discussion of this and related problems see [Sankoff & Kruskal, 1983].
We summarize the dynamic programming formulae used by MODELLER to
obtain the optimal alignment since they differ slightly from those
already published [Sellers, 1974,Gotoh, 1982]. The recursive dynamic
programming formulae that give a matrix ![]() are:
are:
The arrays ![]() ,
, ![]() and
and ![]() are initialized as follows:
are initialized as follows:
The minimal score ![]() is obtained from
is obtained from
Automatic builds 2017-07-19