



Next: restraints.unpick_all() unselect
Up: The restraints class: static
Previous: restraints.make() make
Contents
Index
make_distance(atmsel1, atmsel2, aln, spline_on_site, restraint_group, maximal_distance, residue_span_range=(0, 99999), residue_span_sign=True, distance_rsr_model=1, basis_pdf_weight='LOCAL', basis_relative_weight=0.05, spline_dx=0.5, spline_min_points=5, spline_range=4.0, accessibility_type=8, restraint_stdev=(0.1, 1.0), restraint_stdev2=(0.0, 0.0, 0.0), surftyp=1, distngh=6.0, edat=None, io=None)
- Requirements:
- topology & parameters
This command calculates and selects new distance restraints.
See restraints.make() for full details.
Distance restraints are generated for all pairs of atoms 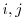 where
atom
where
atom 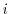 is from selection atmsel1 and atom
is from selection atmsel1 and atom 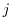 is from selection
atmsel2. Moreover, for a restraint to be created, at least one distance in
the template structures must be less than maximal_distance (in ).
The mean of this basis pdf is equal to the template distance and its standard
deviation
is from selection
atmsel2. Moreover, for a restraint to be created, at least one distance in
the template structures must be less than maximal_distance (in ).
The mean of this basis pdf is equal to the template distance and its standard
deviation 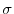 is calculated from an analytic model specified by
distance_rsr_model. Use model 5 for
is calculated from an analytic model specified by
distance_rsr_model. Use model 5 for
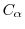 -
distances and model
6 for N-O distances. For models 1 through 6, this standard deviation is
transformed by
-
distances and model
6 for N-O distances. For models 1 through 6, this standard deviation is
transformed by
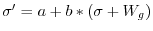 where
where 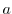 and
and 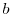 are given
by restraint_stdev and
are given
by restraint_stdev and 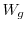 is a gap weighting function of the form
is a gap weighting function of the form
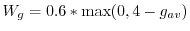 .
. 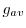 is the average distance of the
two residues involved in the restraint from a gap. For models 3 through 6,
this is additionally transformed by
is the average distance of the
two residues involved in the restraint from a gap. For models 3 through 6,
this is additionally transformed by
![$ \sigma'' = \sigma' + \sum_{i}[d+e*\max(0,f-g_{i})]$](img72.png) where the sum is over each
of the atoms
involved in the distance,
where the sum is over each
of the atoms
involved in the distance, 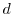
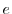 and
and 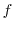 are given by
restraint_stdev2, and
are given by
restraint_stdev2, and 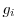 is the distance of each residue from a gap.
The first six models are polynomials and depend on several structural features
of the template and its similarity to the target. The polynomial
coefficients are specified in library file '$PARAMS_LIB'. When
``polynomial model'' 7 is selected, the standard deviation of restraints
is set to constant
. Each basis pdf in the distance pdf corresponds
to one template structure with an equivalent distance.
is the distance of each residue from a gap.
The first six models are polynomials and depend on several structural features
of the template and its similarity to the target. The polynomial
coefficients are specified in library file '$PARAMS_LIB'. When
``polynomial model'' 7 is selected, the standard deviation of restraints
is set to constant
. Each basis pdf in the distance pdf corresponds
to one template structure with an equivalent distance.
In addition, the atom pairs restrained by homology-derived restraints
must by default not be in a chemical bond, chemical angle, dihedral angle,
or on an excluded pairs list. This behavior can be changed by resetting
energy_data.excl_local (see conjugate_gradients()).
Next: restraints.unpick_all() unselect
Up: The restraints class: static
Previous: restraints.make() make
Contents
Index
Ben Webb
2008-02-01