FILE =
 |
'default' |
partial or complete filename |
| DIRECTORY =
|
'' |
directory list (e.g., 'dir1:dir2:dir3:./:/') |
ALIGN_CODES2 =
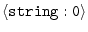 |
'all' |
align codes for alignment2 |
| ALIGNMENT_FORMAT =
|
'PIR' |
format of the alignment file: 'PIR' | 'PAP' | 'QUANTA' | 'INSIGHT' | 'FASTA' |
REMOVE_GAPS =
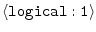 |
on |
whether to remove all-gap positions in input alignment |
STOP_ON_ERROR =
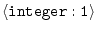 |
1 |
whether to stop on error |