Introduction¶
This document will describe how to get started using ModPipe. If you have more questions, contact Ben in the Sali lab.
Getting ModPipe¶
Sali lab users¶
The best way to use ModPipe is to use one of the stable versions. These
are available as modules, so just run module load modpipe which will
set the MODPIPE environment variable. You can then simply run the main
ModPipe script with:
${MODPIPE}/main/ModPipe.pl
If you want to add new functionality to ModPipe, or you simply want your own copy for some reason, you can check it out from the Sali Lab Subversion server. It is recommended that you use the stable releases, however, unless you’re sure you know what you’re doing.
Users outside the Sali lab¶
Download the ModPipe .tar.gz file and extract it in a suitable directory.
Next, you may need to download and install some external programs needed by
ModPipe (see Installing external programs). Finally, run the setup.py script in
the top-level ModPipe directory with your desired version of Python
(e.g. python3 setup.py or python2 setup.py).
ModPipe is then ready to use by running
modpipe in the bin directory (you may want to add this directory
to your PATH). To be sure it is working correctly, run the test cases by
typing make in the tests directory.
ModPipe methodology¶
Standard ModPipe (Sequence based)¶
ModPipe automates all four steps of the comparative modeling procedure, namely, fold assignment, sequence-structure alignment, model building, and model evaluation. For the sake of efficiency, the fold assignment methods and sequence-structure alignment methods have been combined. ModPipe starts with an input sequence and performs the following steps:
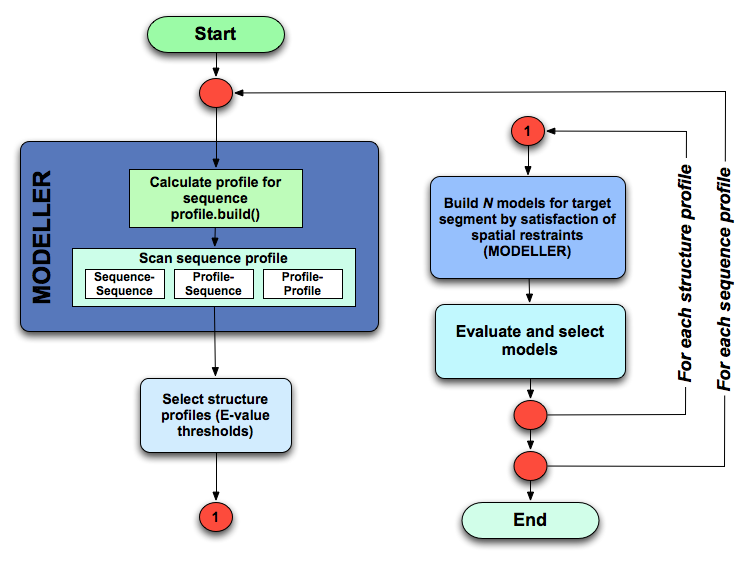
Flowchart depicting the ModPipe protocol.¶
Calculates a profile (multiple sequence alignment) using the
Profile.build()method in Modeller and/or PSI-BLAST.Calculates fold-assignments and sequence-structure alignments using several optional variations of sequence-sequence, profile-sequence, sequence-profile, profile-profile, sequence-consensus methods. All these methods are implemented in Modeller.
Hits (input sequence to template structure matches) with an E-value better than a specified cut-off value are selected.
The selected hits/alignments are clustered to remove redundancy using specified thresholds.
A specified number of models are calculated for each selected hit/alignment. If multiple models are calculated, the best model (for a given hit/alignment) can be selected by one of the many model assessment scores implemented in Modeller.
If an option is specified, small ligands and associated water molecules present in the template structure file can be inherited into the model during model building.
All the calculated models are then subjected to several fold assessment tests. Some of these scores are part of Modeller and some are specific to ModPipe.
The resulting profiles, alignments, models and all scores are written out to files in specific locations within the ModPipe filesystem hierarchy.
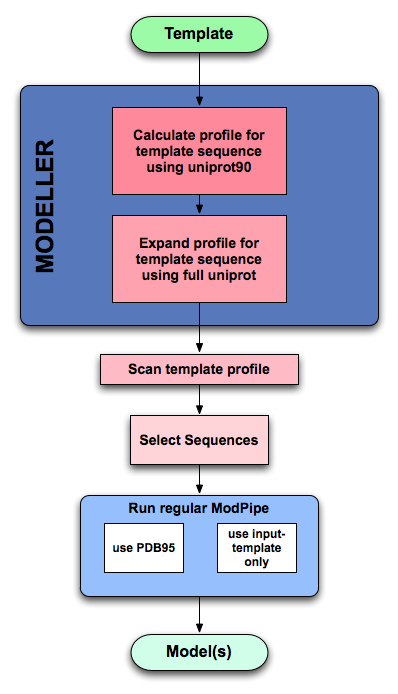
Template Based ModPipe¶
Additionally to the standard ModPipe protocol, a wrapper around it allows the user to run ModPipe in a template based mode. The input is a template PDB structure. ModPipe then creates a profile, collects the sequences in the profile, and modeles those in a standard ModPipe run using the full template library and/or the input template.
ModPipe filesystem or directory hierarchy¶
ModPipe follows a special directory hierarchy to facilitate loading of models into ModBase and for easier bookkeeping.
Every input (target) sequence to ModPipe is given a unique identifier, referred to in this document as a sequence ID. This is a 40-character alphanumeric string; the first 32 characters of this string are the MD5 digest of the full primary sequence (without the header), and the remainder are the first four and last four characters of the sequence. Here is a sample ID string:
97d910a766e44b11cb232a8b78312180MNIFYKNL
There is a root data directory (the ModPipe “repository”) for all data calculated by ModPipe. This directory is specified by the DATDIR variable in the configuration file. In this root directory each sequence processed by ModPipe will have its own sub-directory, which is identified as in this example:
{DATDIR}/97d/97d910a766e44b11cb232a8b78312180MNIFYKNL/
The first sub-directory is the first three characters of the sequence ID; the sub-sub-directory is the full sequence ID.
Each sequence directory will have three sub-directories:
/sequence: contains the sequence, the profile of the sequence, the fold assignments and a summary of all the models calculated.
/alignments: contains all the alignments calculated for this sequence by ModPipe.
/models: contains all the models calculated for all the alignments by ModPipe.
In summary, the data will be stored according to the following (sample) hierarchy:
<DATDIR>/97d/97d910a766e44b11cb232a8b78312180MNIFYKNL/sequence
/alignments
/models
Unique mapping file¶
A unique mapping file (usually given the .unq extension) is placed at the top of the ModPipe filesystem. This file maps the ModPipe sequence IDs to FASTA IDs. Note that one ModPipe identifier may map to multiple FASTA IDs if the input alignment files contain duplicated sequences.
The ModPipe source and its components¶
The source distribution of ModPipe has the subdirectories described below.
/maincontains several scripts that run the high-level ModPipe protocol.
/srccontains utility several scripts that are used by ModPipe for its functionality. These scripts can be used in a stand-alone mode and are not dependent on the special directory hierarchy maintained by ModPipe. Essentially, one could treat them as front-ends for various commands in Modeller.
BuildProfile.py is a front-end for the
Profile.build()method of Modeller. Can be used to calculate profiles for any sequence using command line options.HitsSeqSeq.pl will fetch sequence-structure relationships based on pairwise Smith-Waterman alignments between the query sequence and a database of PDB sequences. It calls
Profile.build()for its functionality.HitsPrfSeq.pl will use a pre-calculated sequence profile to get profile-sequence hits against the database of PDB sequences. Again uses
Profile.build()for its functionality.HitsPrfPrf.pl will calculate profile-profile alignments using a target-profile against a database of structure profiles. It calls the
Profile.scan()command of Modeller for its functionality.MakeModels.pl will take an alignment and produce one or more models based on that alignment.
MergePairwiseToMultiple.pl creates a multiple alignment from a set of pairwise alignments.
AddSeq.py is a flexible tool to assign a single FASTA/PIR file of sequences into a ModPipe directory hierarchy.
CheckProfiles.pl can be used to check the completion of profiles when calculating a large number on the cluster.
GetFullSeqs.pl uses a configuration file. extends or updates a profile and harvests the full-length sequences from a sequence database (tested using uniprot)
/libcontains all the Python and Perl libraries used by ModPipe and other scripts.
/auxcontains miscellaneous scripts to do everyday chores. For instance, CheckPDB.pl checks a PDB file for the number of chains, number of backbone atoms for each chain, resolution, and the number of chain-breaks, etc.
/extcontains various external programs used by ModPipe. These programs are precompiled for use on common architectures. (We cannot distribute these outside of the Sali lab; see Installing external programs for install instructions.)
blast- NCBI’s Basic Local Alignment Search Tool
cd-hitclusters sequences, e.g. from PDB
ce- 3D protein structure comparison by Combinatorial Extension
Jess- binding site similarity checks
mod- Modeller, for alignment, model building, assessment
procheck- checks the stereochemical quality of a protein structure
seg- removes low-complexity regions
spasm- compare 3D motifs
surfnet- cavity volumes
TM-Align- pairwise structure comparisons
Note
All the fold-assignment commands, HitsSeqSeq.py, HitsPrfSeq.py and HitsPrfPrf.pl will produce alignments that are in a format that are ready for modeling. That is, the template chains in the alignments are verified against PDB files for residue numbering and chain identifiers etc. It is more convenient to use these scripts than to use Modeller directly to avoid dealing with re-numbering the alignment files.