This command reads the parameters from the parameter library
given by file, such as the CHARMM 22 parameter file for proteins with
all atoms [MacKerell et al., 1998]. The parameters are added to any already in
memory. This file contains the values for bond lengths, angles,
dihedral angles, improper dihedral angles, and non-bonded interactions.
MODELLER relies on slightly modified CHARMM-22 parameters to reproduce the
protein geometry in the MODELLER environment. For example, for the
default non-hydrogen atoms model, the 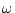 dihedral angle restraints
are stronger than the original CHARMM 22 values which apply to
the all-hydrogen model. For a sparse discussion of the parameter
library, see the FAQ Section 1.8, Question 10.
dihedral angle restraints
are stronger than the original CHARMM 22 values which apply to
the all-hydrogen model. For a sparse discussion of the parameter
library, see the FAQ Section 1.8, Question 10.
Note that, in contrast to older versions of MODELLER, the non-bonded spline
parameters used in loop modeling are not read by this function. See instead
the documentation for the separate group_restraints class, in
section 3.8, for more information.