



Next: Comparative modeling class reference
Up: MODELLER updates
Previous: Changes since release 9v8
Contents
Index
- Residues treated as BLK no longer have to be HETATM residues; they will
be marked as ATOM if so defined in restyp.lib or in the template PDB
(for example, DNA or RNA residues). automodel.nonstd_restraints() now
builds restraints on both HETATM and BLK residues.
- Python methods that previously expected lists or tuples can now work with
other iterables too (such as generators). For example, in Python 2.4 you
can now say 's = selection(x for x in mdl.atoms where x.biso > 0.5)' to
select all atoms with B
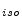 over 0.5.
over 0.5.
- The file argument to model.write_data() is now optional, since we
may only want the calculated property in B
, and some properties (e.g. CRV)
cannot be written to files anyway.
- Bonds between BLK residues are now detected by a simple distance criterion
and are restrained more strongly than other distances by
automodel.nonstd_restraints().
- A new method Chain.join() allows chain breaks between chains to
be removed.
- Files compressed with 'xz' can now be read, provided that the
'xz' binary is available on your system.
- Bundled version of HDF5 updated to 1.8.3.
- The allow_alternates option to alignment.append() will now
allow G in the alignment sequence to match any non-standard residue type
in the PDB file which maps to G. This is useful if the alignment sequence
is 'cleaned' as by sequence_db.read().
- When using GB/SA with BLK residues, each BLK atom will now be treated as
an uncharged particle with a small radius, rather than causing an error.
- In sequence_db.filter(), if two sequences fail to align, don't
terminate the entire run; rather, place them in separate clusters.
- density.read() now supports CCP4 files, and reads the map and
voxel sizes from the density map file header if not specified by the user.
- Bugfix: invalid PAP files no longer crash Modeller with an assertion
failure.
- Bugfix: projected coordinates from environ.principal_components()
are now written correctly to the output file.
- Bugfix: models can now be built containing HSE or HSP residues without
the program terminating with a cryptic MDT error; homology-derived
restraints for these residues will look very similar to those for HIS.
- Bugfix: assigning to Residue.num now gives a correctly-formatted
(right-aligned) PDB residue number by default.
- Bugfix: assigning to Residue.code, Residue.name or
Residue.pdb_name, where the residue is in an alignment, now works correctly
(previously, the new one-letter code was not used when the alignment was
written to disk).
- Bugfix: mainchain curvature values for terminal residues were incorrect
in some cases.
- Bugfix: mainchain curvature calculation no longer spans chain breaks.
- Bugfix: in profile.scan(), if the target profile has no name, do not
trigger a GLib CRITICAL error.
- Bugfix: automodel with write_intermediates=True now works
correctly.
Next: Comparative modeling class reference
Up: MODELLER updates
Previous: Changes since release 9v8
Contents
Index
Automatic builds
2011-03-29