


 Download files
Download files Verified to work with the
Verified to work with the  To install the software needed to reproduce this system with the
To install the software needed to reproduce this system with the
 To set up the environment on the UCSF Wynton cluster to run
this system, run:
To set up the environment on the UCSF Wynton cluster to run
this system, run:
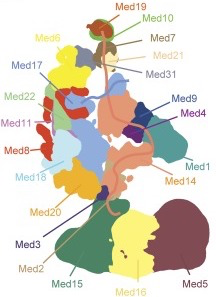
The 21-subunit Mediator complex transduces regulatory information from enhancers to promoters, and performs an essential role in the initiation of transcription in all eukaryotes. In this study we performed chemical cross-linking and mass spectrometry, and combined the results with information from X-ray crystallography, homology modeling, and cryo-electron microscopy by an integrative modeling approach to determine a 3-D model of the entire Mediator complex. The approach is validated by the use of X-ray crystal structures as internal controls and by consistency with previous results from electron microscopy and yeast two-hybrid screens. The model shows the locations and orientations of all Mediator subunits, as well as subunit interfaces and some secondary structural elements. Segments of 20-40 amino acid residues are placed with an average precision of 20 Å. The model reveals roles of individual subunits in the organization of the complex.
The scripts were run for the publication with IMP.pmi version from 915c00bac191ab32f022ae425facf538b64e3e54 to 32a774583007c3a7135c8adf970846ea7a52f453 and IMP version 3d7d35a367342a94655b5ca2f75cc4d4fb2d5c71. They should also
work with any recent version of IMP.
For more details on how to install IMP, run the modeling scripts and analyze the results using IMP and IMP.pmi see the IMP tutorial.
In brief, to use Replica Exchange, IMP must be compiled using an openmpi c++ compiler. We suggest to compile openmpi with the --disable-dlopen flag. See the IMP building instructions.
To run the modeling script, access the sampling/modeling directory
and then run with 64 threads (64 replicas):
mpirun -np 64 python modeling.py
analysis: analysis scripts and clustering results of the whole ensemble of solutions, the two ensemble halves, and the jackknifing
sampling: sampling and modeling scripts, all input data
clustering: the clustering results for the whole ensemble of solutions
clustering_half1: the clustering results for the ensemble of solutions considering the first half of all models produced
clustering_half2: the clustering results for the ensemble of solutions considering the second half of all models produced
clustering_jackknifing: the clustering results for the ensemble of solutions obtained jackknifing the cross-link dataset
present in all analysis/clustering* directories
clustering.py: clustering script
kmeans_weight_500_1: the set of 500 best scoring models collected in a single cluster
kmeans_weight_500_4: the set of 500 best scoring models divided into 4 clusters
content of kmeans_weight_500_4 directory
cluster.0: data for cluster 1
cluster.1: data for cluster 2
cluster.2: data for cluster 3
cluster.3: data for cluster 4
precision.0.0.out,precision.0.1.out,precision.0.2.out,precision.0.3.out ... precision.3.3.out: files containing the precision of a cluster (i.e., the files with the same indexes, precision.i.i.out, e.g., precision.0.0.out) and the files containing the distance between the clusters (i.e., files with different indexes, precision.i.j.out).
content of cluster.* directories
0.pdb,1.pdb,2.pdb....: the pdb files of the solutions
0.rmf3,1.rmf3,2.rmf3,...: the rmf files of the solution (can be opened with UCSF Chimera)
rmsf.med10.dat,rmsf.med11.dat,...: text file of the RMSF analysis
rmsf.med10.pdf,rmsf.med11.pdf,...: pdf file of the RMSF analysis
show_localization.py: chimera session file to display the localization densities
stat.out: stat file containing all relevant information on the score, etc.
view1_matrix: used by Chimera session file.
view2_matrix: used by Chimera session file.
XL_table_middle.pdf: pdf file of the cross-link map for the middle module
XL_table_tail.pdf: pdf file of the cross-link map for the tail module
present only in analysis/clustering
graph_plotting.py: compute the graph of the interactions between the subunits
high_confidence_structure.py: get the cluster-center solution and map the RMSF of beads with a color. Need Chimera to display it.
precision_rmsf.py: calculate the precision of clusters, their mutual distance and the RMSF
XL_table_middle.py: calculate the contact map and the cross-link map of the middle module
XL_table_tail.py: calculate the contact map and the cross-link map of the tail module
CXMS_files: cross-link datasets
em_map_files: input electron microscopy maps, with their GMM representation
fasta_files: fasta files with primary sequences of Mediator subunits
model_gmm_files: GMM representation of Mediator domains
modeling: modeling script
pdb_files: crystallographic structures and homology models
full_med_splitmods.txt: mediator cross-link dataset, cvs file.
jackknife_analysis: contains jackknifed datasets
asturias_mediator.mrc: the input file from Asturias lab EMD-2634
asturias_mediator_translated.mrc: the map translated in the coordinate center.
asturias_middle_module_translated.mrc: the middle module density
asturias_middle_module_translated_resampled.mrc: resampled middle module.
asturias_middle_module_translated_resampled.mrc.gmm.29.mrc: mrc file with the GMM approximation of the middle module density, with 29 gaussians.
asturias_middle_module_translated_resampled.mrc.gmm.29.txt: text file with 29 gaussian coordinates and covariance tensors for the middle module.
asturias_tail_module_translated.mrc: tail module density.
asturias_tail_module_translated_resampled.mrc: resampled tail module.
asturias_tail_module_translated_resampled.mrc.gmm.49.mrc: mrc file with the GMM approximation of the tail module density, with 49 gaussians.
asturias_tail_module_translated_resampled.mrc.gmm.49.txt: text file with 49 gaussian coordinates and covariance tensors for the tail module.
calculate.density.sh: script used to calculate the GMMs.
cr_mid_fullmed10.pdb: middle module pdb
head_module_em_aligned_translated.pdb: head module pdb
med16.NTD.phyre.model.pdb: homology model of med16
Author(s): Riccardo Pellarin, Charles Greenberg
Date: September 24th, 2015
License: CC-BY-SA-4.0. This work is freely available under the terms of the Creative Commons Attribution-ShareAlike 4.0 International License.
Last known good IMP version:
Publications:
- P. Robinson, M. Trnka, R. Pellarin, C. Greenberg, D. Bushnell, R. Davis, A. Burlingame, A. Sali, R. Kornberg. Molecular architecture of the yeast Mediator complex, eLife 4, e08719, 2015