Tutorial
Advanced example:
Modeling of a protein-ligand complex based on multiple templates and users
specified restraints
All input and output files for this example are available to download,
in either zip format (for Windows) or
.tar.gz format (for Unix/Linux).
An important aim of modeling is to contribute to understanding of the
function of the modeled protein. Inspection of the
4mdh:A template structure (built in the
basic modeling tutorial) revealed that loop 93-100,
one of the functionally most important part of the enzyme, is more disordered
than the rest of the protein. The long active site loop appears to be flexible
in the absence of a ligand and could not be seen well in the diffraction map.
The unreliability of the template coordinates and the inability of
MODELLER to model long insertions is why this loop
was poorly modeled in TvLDH, as indicated by
PROSAII.
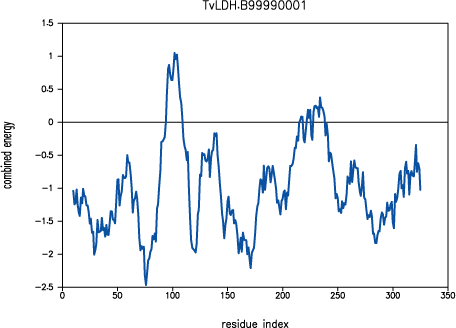
PROSAII profile for model TvLDH.B99990001
Since we are interested in understanding differences in specificity between
two similar proteins, we need to build precise and accurate models. Therefore,
we need to search for another template malate dehydrogenase structure, which
may have a lower overall sequence similarity to TvLDH, but a better resolved
active site loop. The old and new templates can then be used together to get a
model of TvLDH. The active site loop tends to be more defined if the structure
is solved together with its physiological ligand and a co-factor. The model
based on a template with ligands bound is also expected to be more relevant
for the purposes of our study of enzymatic specificity, especially if we also
build the model with the ligands.
1emd, a malate dehydrogenase from E. coli,
was identified in PDB. While the 1emd sequence shares
only 32% sequence identity with TvLDH, the active site loop and its environment
are more conserved. The loop in the 1emd structure is
well resolved. Moreover, 1emd was solved in the
presence of a citrate substrate analog and the NADH cofactor. The new alignment
in the PAP format is shown below (file
`TvLDH-4mdh-1emd_ed.pap').
_aln.pos 10 20 30 40 50 60
1emd_ed --------------------------------------------------------------------
4mdhA -SEPIRVLVTGAAGQIAYSLLYSIGNGSVFGKDQPIILVLLDITPMMGVLDGVLMELQDCALPLLKDV
TvLDH MSEAAHVLITGAAGQIGYILSHWIASGELYG-DRQVYLHLLDIPPAMNRLTALTMELEDCAFPHLAGF
_consrvd
_aln.p 70 80 90 100 110 120 130
1emd_ed ------------------SAGVRRKPGMDRSDLFNV--------------NAGI--------------
4mdhA IATDKEEIAFKDLDVAILVGSM--------------PRRDGMERKDLLKANVKIFKCQGAALDKYAKK
TvLDH VATTDPKAAFKDIDCAFLVASMPLKPGQVRADLISS--------------NSVIFKNTGEYLSKWAKP
_consrvd * *
_aln.pos 140 150 160 170 180 190 200
1emd_ed --------------------------------------------------------------------
4mdhA SVKVIVVGNPANTNCLTASKSAPSIPKENFSCLTRLDHNRAKAQIALKLGVTSDDVKNVIIWGNHSST
TvLDH SVKVLVIGNPDNTNCEIAMLHAKNLKPENFSSLSMLDQNRAYYEVASKLGVDVKDVHDIIVWGNHGES
_consrvd
_aln.pos 210 220 230 240 250 260 270
1emd_ed --------------------------------------------------------------------
4mdhA QYPDVNHAKVKLQAKEVGVYEAVKDDSWLKGEFITTVQQRGAAVIKARKLSSAMSAAKAICDHVRDIW
TvLDH MVADLTQATFTKEGKTQKVVDVLDHD-YVFDTFFKKIGHRAWDILEHRGFTSAASPTKAAIQHMKAWL
_consrvd
_aln.pos 280 290 300 310 320 330 340
1emd_ed --------------------------------------------------------------------
4mdhA FGTPEGEFVSMGIISD-GNSYGVPDDLLYSFPVTIK-DKTWKIVEGLPINDFSREKMDLTAKELAEEK
TvLDH FGTAPGEVLSMGIPVPEGNPYGIKPGVVFSFPCNVDKEGKIHVVEGFKVNDWLREKLDFTEKDLFHEK
_consrvd
_aln.pos 350 360 370 380 390 400
1emd_ed ----------VKNLVQQVAKTCPKACIGIITNPVNTTVAIAAEVLKKAGVYDKNKLFGVTTLDIIRSN
4mdhA ETAFEFLSSA----------------------------------------------------------
TvLDH EIALNHLAQ-----------------------------------------------------------
_consrvd
_aln.p 410 420 430 440 450 460 470
1emd_ed TFVAELKGKQPGEVEVPVIGGHSGVTILPLLSQVPGVSFTEQEVADLTKRIQNAGTEVVEAKAGGGSA
4mdhA --------------------------------------------------------------------
TvLDH --------------------------------------------------------------------
_consrvd
_aln.pos 480 490 500 510 520 530 540
1emd_ed TLSMGQAAARFGLSLVRALQGEQGVVECAYVEGDGQYARFFSQPLLLGKNGVEERKSIGTLSAFEQNA
4mdhA --------------------------------------------------------------------
TvLDH --------------------------------------------------------------------
_consrvd
_aln.pos 550 560
1emd_ed LEGMLDTLKKDIALGQEFVNK/-..
4mdhA ---------------------/.--
TvLDH ---------------------/..-
_consrvd
File: TvLDH-4mdh-1emd_ed.pap
The modified alignment refers to an edited 1emd
structure (1emd_ed), as a second template. The
alignment corresponds to a model that is based on
1emd_ed in its active site loop and on
4mdh:A in the rest of the fold. Four residues on both
sides of the active site loop are aligned with both templates to ensure that
the loop has a good orientation relative to the rest of the model.
The modeling script below has several changes with respect to
`model-single.top'. First, the name of the
alignment file assigned to ALNFILE is updated. Next, the
variable KNOWNS is redefined to include both templates.
Another change is an addition of the `SET HETATM_IO = ON'
command to allow reading of the non-standard pyruvate and NADH residues from
the input PDB files. The script is shown next (file
`model-multiple-hetero.top').
INCLUDE
SET ALNFILE = 'TvLDH-4mdh-1emd_ed.ali'
SET KNOWNS = '4mdhA' '1emd_ed'
SET SEQUENCE = 'TvLDH'
SET STARTING_MODEL = 1
SET ENDING_MODEL = 5
SET HETATM_IO = ON
CALL ROUTINE = 'model'
SUBROUTINE ROUTINE = 'special_restraints'
ADD_RESTRAINT ATOM_IDS = 'NH1:161' 'O1A:334',;
RESTRAINT_PARAMETERS = 2 1 1 22 2 2 0 3.5 0.1
ADD_RESTRAINT ATOM_IDS = 'NH2:161' 'O1B:334',;
RESTRAINT_PARAMETERS = 2 1 1 22 2 2 0 3.5 0.1
ADD_RESTRAINT ATOM_IDS = 'NE2:186' 'O2:334',;
RESTRAINT_PARAMETERS = 2 1 1 22 2 2 0 3.5 0.1
RETURN
END_SUBROUTINE
File: model-multiple-hetero.top
A ligand can be included in a model in two ways by
MODELLER. The first case corresponds to the ligand
that is not present in the template structure, but is defined in the
MODELLER residue topology library. Such ligands
include water molecules, metal ions, nucleotides, heme groups, and many other
ligands (see question 17 in the
the MODELLER FAQ). This situation is not explored
further here. The second case
corresponds to the ligand that is already present in the template structure. We
can assume either that the ligand interacts similarly with the target and the
template, in which case we can rely on MODELLER to
extract and satisfy distance restraints automatically, or that the relative
orientation is not necessarily conserved, in which case the user needs to
supply restraints on the relative orientation of the ligand and the target (the
conformation of the ligand is assumed to be rigid). The two cases are
illustrated by the NADH cofactor and pyruvate modeling, respectively. Both NADH
and cofactor are indicated by the `.' characters at the end of each sequence in
the alignment file above (the `/' character indicates a chain break). In
general, the `.' character in MODELLER indicates an
arbitrary generic residue called a ``block'' residue (for details see the
section on block
residues in the MODELLER manual). The
1emd structure file contains a
citrate substrate analog. To obtain a model with pyruvate, the physiological
substrate of TvLDH, we convert the citrate analog in
1emd into pyruvate by deleting the group
CH(COOH)2, thus obtaining the
1emd_ed template file. A major advantage of using the
`.' characters is that it is not necessary to define the residue topology.
To obtain the restraints on pyruvate, we first superpose the structures of
several LDH and MDH enzymes solved with ligands. Such a comparison allows to
identify absolutely conserved electrostatic interactions involving catalytic
residues Arg161 and His186 on one hand, and the oxo groups of the lactate and
malate ligands on the other hand. The modeling script can now be expanded by
appending a routine that specifies the user defined distance restraints
between the conserved atoms of the active site residues and their substrate.
The ADD_RESTRAINT command has two arguments.
ATOM_IDS defines the restrained atoms, by specifying their
atom types and the residue numbers as listed in the model coordinate file.
RESTRAINT_PARAMETERS defines the restraints, by specifying
the mathematical form (e.g., harmonic, cosine, cubic spline), modality, the
type of the restrained feature (e.g., distance, angle, dihedral angle), the
number of atoms in the restraint, and the restraint parameters. In this case,
a harmonic upper bound restraint of 3.5±0.1 is imposed on the distances
between the specified pairs of atoms. A trick is used to prevent
MODELLER from automatically calculating distance
restraints on the pyruvate-TvLDH complex; the ligand in the
1emd_ed template is moved beyond the upper bound on
the ligand-protein distance restraints (i.e., 10).
The new script produces a model with a significantly improved
PROSAII profile. The predicted error in the 90-100
active site loop is much less and practically resolved in the loop region
220-250.
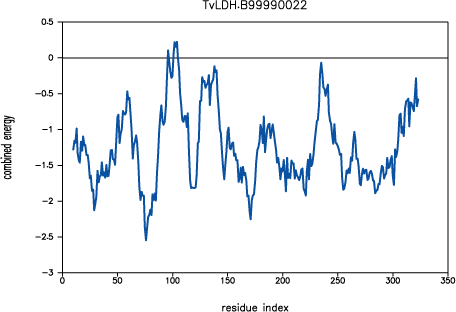
PROSAII profile for model TvLDH.B99990022
The overall Z-score is improved from -10.7 to -11.7, which compares well
with the template Z-score of -12.7. With this favorable evaluation, we gain
confidence in the final model. The model was used for interpreting
site-directed mutagenesis experiments aimed at elucidating the determinants of
enzyme specificity in this class of enzymes.
|