

Tutorial
Iterative example:
The alignment-modeling-evaluation cycle. The case of the
Haloferax volcanii dihydrofolate reductase.
All input and output files for this example are available to download, in either zip format (for Windows) or .tar.gz format (for Unix/Linux).
Several structures of dihydrofolate reductase (DHFR) are known. However, the structure of DHFR from Haloferax volcanii was not known and its sequence identity with DHFRs of known structure is rather low at ~30%. A model of H. volcanii DHFR (HVDFR) was constructed before the experimental structure was solved. This example illustrates the power of the iterative alignment-modeling-evaluation approach to comparative modeling.
Of all the available DHFR structures, HVDHFR has the sequence most similar to DHFR from E. coli. The PDB entry 4dfr corresponds to a high resolution (1.7Å) E. coli DHFR structure. It contains two copies of the molecule, named chain A and chain B. According to the authors, the structure for chain B is of better quality than that of chain A. The following script file aligns HVDFR and chain B of 4dfr.
from modeller import *
env = environ()
mdl = model(env, file='4dfr.pdb', model_segment=('FIRST:B', 'LAST:B'))
aln = alignment(env)
aln.append_model(mdl, align_codes='4dfr')
aln.append(file='hvdfr.seq', align_codes='hvdfr')
aln.align2d()
aln.write(file='hvdfr-4dfr.ali')
aln.write(file='hvdfr-4dfr.pap', alignment_format='PAP',
alignment_features='INDICES HELIX BETA')
File: align2d-4.py
Some options used in this example include model_segment, which is used to indicate chain B of 4dfr; and alignment_features, which is used to output information such as secondary structure to the alignment file in the PAP format.
_aln.pos 10 20 30 40 50 60 4dfr -MISLIAALAVDRVIGMENAMPW-NLPADLAWFKRNTLDKPVIMGRHTWESIGRPLPGRKNIILSSQP hvdfr MELVSVAALAENRVIGRDGELPWPSIPADKKQYRSRIADDPVVLGRTTFESMRDDLPGSAQIVMSRSE _helix 999999999999 999999999 _beta 9999999999 999999 99999999 _aln.p 70 80 90 100 110 120 130 4dfr GTDDRVTWVKSV----DEAIAACGDVPEIMVIGGGRVYEQFLPKAQKLYLTHIDAEVEGDTHFPDYEP hvdfr RSFSVDTAHRAASVEEAVDIAASLDAETAYVIGGAAIYALFQPHLDRMVLSRVPGEYEGDTYYPEWDA _helix 99 99999999 99999999 _beta 99999 9999999 999999999 _aln.pos 140 150 160 4dfr DDWESVFSEFHDADAQNSHSYCFKILERR hvdfr AEWELDAETDHEG---FTLQEWVRSASSR _helix _beta 999999999999 999999999999
File: hvdfr-4dfr.pap
Using the PIR alignment file hvdfr-4dfr.ali, an initial model is calculated:
from modeller import *
from modeller.automodel import *
env = environ()
a = automodel(env, alnfile='hvdfr-4dfr.ali',
knowns='4dfr', sequence='hvdfr')
a.starting_model = 1
a.ending_model = 1
a.make()
File: model4.py
Because the sequence identity between 4dfr and HVDFR is relatively low (30%), the automated alignment is likely to contain errors. The evaluation of the model with the DOPE potential in MODELLER shows two regions with positive energy.
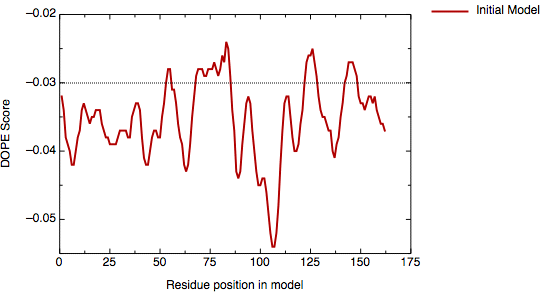
DOPE score profile for model inihvdfr.B99990001.pdb
The first region is around residue 85, the second region is at the C-terminal end of the protein. Referring to the target--template alignment above (hvdfr-4dfr.pap), it is easy to understand why the first positive peak appears. The insertion around position 85 of the alignment was placed in the middle of an α-helix in the template (the "9" characters on the first line below the sequence mark the helices). Moving the insertion to the end of the α-helix may improve the model.
The second problem, which occurs in the C-terminal region of the alignment, is less clear. The deletion in that region of the alignment corresponds to the loop between the last two β-strands of 4dfr (a β-hairpin). Since the profile suggests that this region is in error, an alternative alignment should be tried. One possibility is that the deletion is actually longer, making the C-terminal β-hairpin shorter in HVDFR. One plausible alignment based on these considerations is shown here.
_aln.pos 10 20 30 40 50 60 4dfr M-ISLIAALAVDRVIGMENAMPW-NLPADLAWFKRNTLDKPVIMGRHTWESIGRPLPGRK hvdfr MELVSVAALAENRVIGRDGELPWPSIPADKKQYRSRIADDPVVLGRTTFESMRDDLPGSA _helix 999999999999 999999999 _beta 9 999999999 999999 999 _aln.pos 70 80 90 100 110 120 4dfr NIILSSQPGT--DDRVTWVKSVDEAIAACG--DVPEIMVIGGGRVYEQFLPKAQKLYLTH hvdfr QIVMSRSERSFSVDTAHRAASVEEAVDIAASLDAETAYVIGGAAIYALFQPHLDRMVLSR _helix 9999999999 99999999 _beta 99999 99999 9999999 9999999 _aln.pos 130 140 150 160 4dfr IDAEVEGDTHFPDYEPDDWESVFSEFHDADAQNSHSYCFKILERR---- hvdfr VPGEYEGDTYYPEWDAAEWELDAETDHE-------GFTLQEWVRSASSR _helix _beta 99 999999999999 999999999999
File: hvdfr-4dfr-2.pap
A new model was calculated using this alignment and the script, modified to use the new alignment (see file `model5.py'). Its DOPE score profile is shown in the next figure.
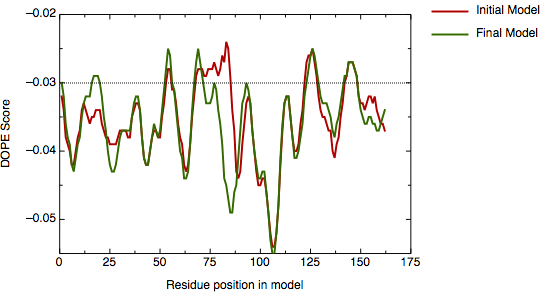
DOPE profile for model hvdfr.B99990001.pdb
The main positive peak disappeared and the new global DOPE score dropped from -15498.7 to -15720.3.
The process outlined here could be iterated until no improvement in the evaluation can be achieved. This iterative alignment-building-evaluation process has been developed in the MOULDER protocol which will be further implemented in MODELLER .